Code Examples¶
Counting microRNAs¶
Requires you provide at least one fastq file and have a short read aligner installed. You should also specify the species being mapped to:
import smallrnaseq as smrna
res = smrna.map_mirbase(files=['test_1.fastq','test_2.fastq'], overwrite=True, aligner='bowtie',
species='hsa', pad5=3, pad3=5)
Counting isomiRs¶
This method is used to count isomiRs using results from previously mapped reads. So a sam file is required:
smrna.count_isomirs(samfile, truecounts, species='bta')
Mapping to the genome¶
This requires a reference genome and a gtf file with miRNA features:
featcounts = srseq.map_genome_features(['test_1.fastq'], 'bos_taurus', gtffile,
outpath='ncrna_map', aligner='subread', merge=True)
Novel miRNA prediction¶
The built-in method for novel prediction should be considered a somewhat ‘quick and dirty’ method at present but is relatively fast and convenient to use. The basic idea is to take clusters of reads that could be mature sequence and find suitable precursors. Structural features of each precursor are then scored using a classifier. The best candidate is selected is there is at least one. We have followed a similar approach to the miRanalyzer method.
The following features are currently used in our algorithm, most are the same as those used in sRNAbench (miRanalyzer). The diagram below may help to clarify some of the terminology used.
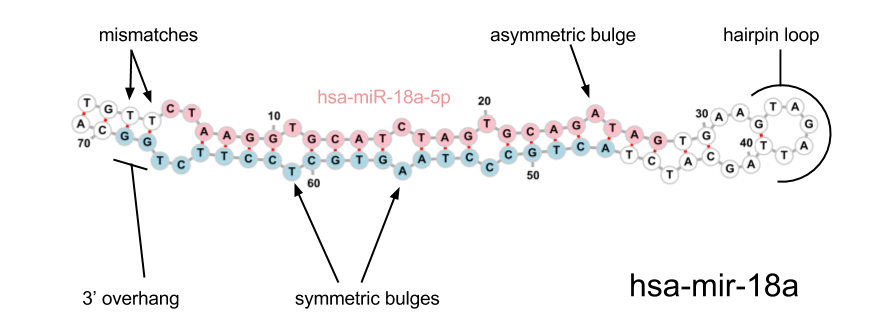
To predict miRNAs you need to have run mapping on genome. Then use the sam file and read counts to get the true reads and input this into the method find_mirnas with a reference genome fasta file. The reference fasta must match the bowtie index you used for alignment:
from smallrnaseq import novel
import pandas as pd
#single file prediction
readcounts = pd.read_csv('countsfile.csv')
samfile = 'mysamfile.sam'
reads = utils.get_aligned_reads(samfile, readcounts)
new = novel.find_mirnas(reads, ref_fasta)
Differential Expression¶
Assuming we have all the raw files, they need to be adapter trimmed. Optionally you can remove other ncrnas before counting your target rnas class, though that may not be advisable. The following code maps all the files to bovine mature miRNAs and counts the mapped genes, then saves the results to a csv file which has the counts in one column per sample. You can skip this if you already have the counts file:
import pandas as pd
import smallrnaseq as smrna
from smallrnaseq import base, utils, de
path = 'pathtodata'
base.BOWTIE_INDEXES = 'bowtie_index'
refs = ['mirbase-bta'] #name of bowtie index
files = glob.glob(path+'/*.fastq')
outpath = 'ncrna_map'
#map to selected annotation files
counts = smrna.map_rnas(files, refs, outpath, overwrite=True)
R = smrna.pivot_count_data(counts, idxcols=['name','db'])
R.to_csv('mirna_counts.csv',index=False)